format_diagnosis.pl¶
To determine what format you have, if it is ambiguous, format_diagnosis.pl may be able to help. The script will output information that you can compare with the table below to see if another format may work for you. To use the script:
perl format_diagnosis.pl [input_file]
The output will look something like this:

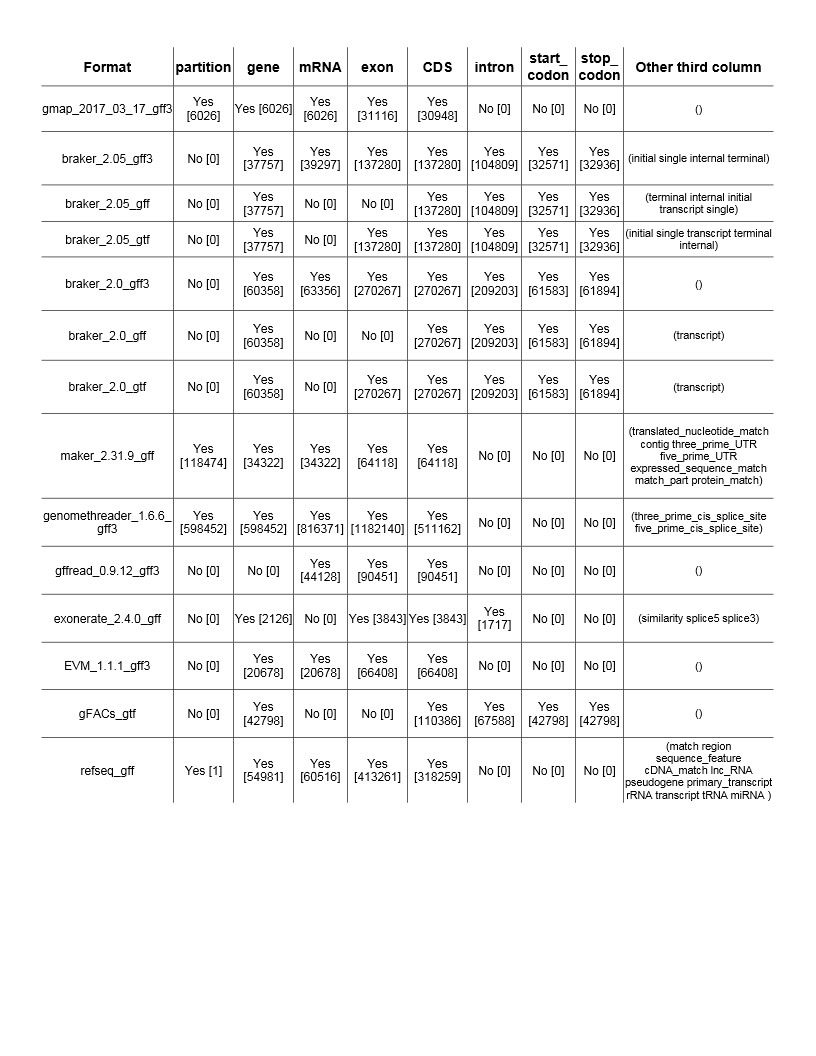
The data tells you what information is present followed by the observed quality of the feature. In the above output, the line of the “gene” feature, comes up 278 times and matches that with mRNA. This is not always the case. It also has exon lines and CDS lines but NOT at the same frequency. So CDS will be the more important feature.
Given the comparison, you could choose several formats that might work. braker_2.05_gff3, braker_2.05_gtf, gFACs_gtf, and several more.
FORMAT properties:
NOTE: In this table, know that each format example is NOT the same file. The information inside the [brackets] is just an example number and judgement on format should be made from ratios. Your file will not fit the actual numbers in the brackets above, but the ratio between a format’s exon to CDS counts may be the same. These were derived from my own collection of sample files across different species and projects.