About¶
gFACs was created during the annotation of the megagenome of loblolly pine. In dealing with gene models created from different softwares and alignment tools, we needed a way to filter and merge these models. Unfortunately, no such system existed so gFACs was designed and developed to fill this niche. The applications that generate gene model evidence include aligners and ab initio gene prediction software. These programs report their predictions and alignments in a similar structured gene transfer format (gtf) or general file format (gff) however there is little consistency across these standards. You can read more about the general structures here. gFACs will filter and select final gene models based upon user provided filters regarding their structural attributes. In addition, gFACs can optionally consider functional annotation from the EnTAP application as an additional filter to define true models.
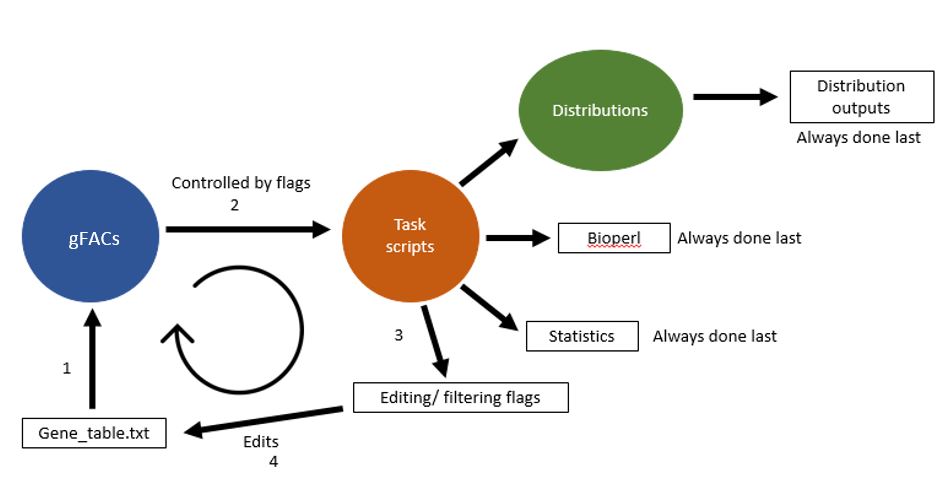
The flow of gFACs.pl is controlled by the master script gFACs.pl. Flags and input files are processed by the master script, and a series of task-specific scripts are called upon to edit and filter gene or alignment models.
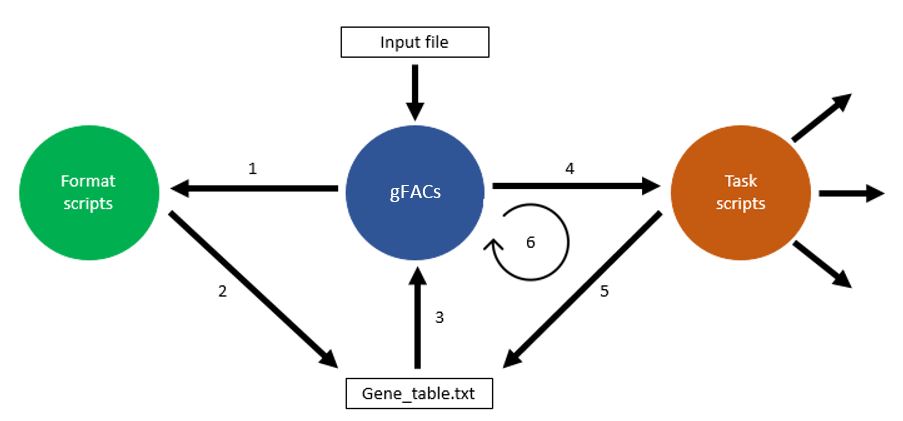
The primary input is an annotation file, either in gene transfer format (gtf) or general feature format (gff/gff3). Since these file types are variable across the applications that generate them, formats designed to fit a particular software’s output must be created. Specific scripts in the folder format_scripts/ are used to convert the input into a median file type called gene_table.txt.The user is able to input their specific file type but they must inform gFACs about the format of the file. For specific formats and how to provide this information, see the supported input section on it.
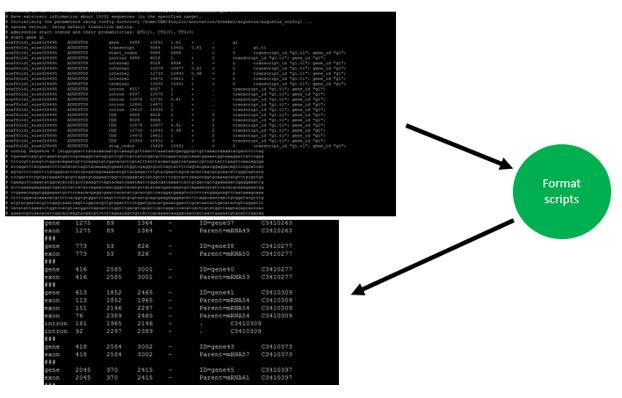
The gene table¶
gene_table.txt (referred to also as the gene table) is specfiic to gFACs and is created to hold the minimum amount of information needed to uniformly apply the filtering options.
The gene table is the most important file for this program as it is used and edited in every step. Each flag or task in evaluation needs the format of the gene table to work successfully. The gene table will always have the gene models or alignments that are retained. Here is an example of gene table format:
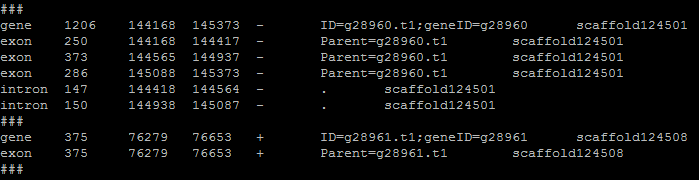
- The columns go:
- Gene part
- Length
- Start
- Stop
- Strand
- ID (8th column from input file)
- Scaffold/chromosome (needed for fasta commands)
Further scripts expect the gene table to be in the output directory called gene_table.txt. If you are using a prefix, it will look for the file with the prefix. The task-scripts also create their own files, notably if things are being separated, such as potential splice variants. However, the retained genes will always be renamed or concatenated into the master gene table.
Intron prediction¶
The gene table provides intron information that is not always found in the input file. Even if the input format does provide introns, they will be recalculated based on the positions of predicted exons. In the format step, a temporary file is created that will terminated upon step completion. The purpose is to add a divider between gene families. The ### line in the gene table allows for a clear break between different genes. The script then revisits the temp file, calculates introns, and makes final formatting shifts.
Here is an example of the temporary file:
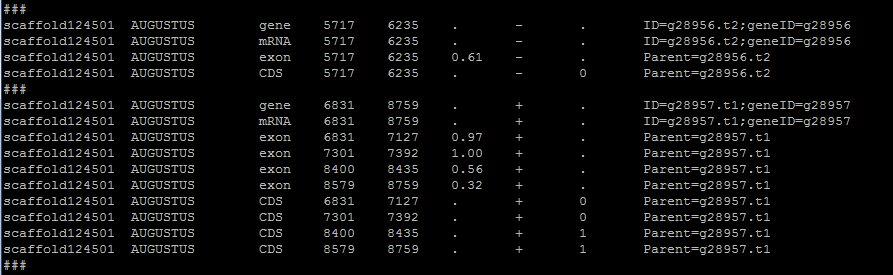
Introns are the sequence between exons. Lengths and start and stop coordinates are calculated based on exon information. To accomplish this, exon lengths are pushed into an array and called by position. This method is more universally reliable but prone to errors involving overlapping exons. This can be resolved with a flag –splice-rescue which I recommend.

Task scripts¶
Once the formatting has completed, and the gene table has been created, filtering as designated by flags is done cyclically on the gene table until the final set of genes is produced. The order of filtering flags is pre-determined although this does not change the final result. For example, whether or not monoexonic genes are removed first or last will not change how many monoexonics appear in the final iteration of the gene table (spoiler alert: none).
Once all filtering is done, other commands are activated that involve processing or analyzing the final sets. This includes statistics, analysis of splice types, or distributions.
The log file¶
In addition to the gene table, the gFACs_log.txt file is created every time the script is run, no exceptions. If a prefix is included, the log will have this prefix. The log file is reported by the master script and is appended with information regarding filtering at each step. It will also report what flags are being activated and the very specific corresponding system commands.
The log may be helpful for the user to see what is happening and the results of a particular filter. It is also helpful for noticing bugs and verifying script efficacy. gFACs supports readable and understandable log files!