Basic flags¶
These are flags that can be run on any input without the addition of a fasta or EnTAP file. Given that, these flags are not capable of performing sequence level analysis. You can include as many flags as you want in any order. However, the order in which the flags are run is predetermined by the gFACs.pl script. This section is designed to tell you what the flags do conceptually. This is not the true order. See the log file for the order as the log is printed in sequence.
-p [prefix]¶
All files created will have your designated prefix. For example, if you provide the prefix “test”, your gene table will be called test_gene_table.txt. Every file (even temporary files) will have this prefix. However, it is not a mandatory argument.
--false-run¶
This flag allows for the user to understand the effect of filters on the original input without an additive effect. Before committing to a gFACs run, it may be important to understand what gFACs is planning on removing from the original set of models. When gFACs filters, it removes sequentially from a shrinking pool of models. However, it may be important to know how many gene models would be removed from the original set. How many overall do not have a stop codon? How many overall are of a certain CDS length?
This flag allows gFACs to run normally but always resets to the original set of genes in the gene table. Therefore, the log will print what is removed and retained but never follows through with filtering. Resulting files will be the gene table completely unedited and the log file will reflect resetting numbers.
NOTE: This flag does NOT keep the results of splice rescue nor unique genes only. This is problematic with overlap when isoforms or multiple RNA evidence models are within the same called gene. Sequence pull filters like in-frame stop codon, fasta creation, or analysis can then be wrong. For example, when setting in-frame stops to 0, all genes with isoforms will print one after another in a long sequence string that will have as many stop codons as models presented. This may create the appearance of increased removal of models. To work around this problem, run classic gFACs on your set using --splice-rescue and --unique-genes-only. Then use --false-run on that parsed gene table output. Feel free to contact me if you have problems!
--no-processing¶
The following occurs by DEFAULT. Use –no-processing to opt out.
Processing involves two scripts: task_scripts/overlapping_exons.pl and task_scripts/splice_variants.pl. The first analyzes the gene table for overlapping exon space and then the second analyzes genes with overlapping exon space for evidence that these are separate transcripts or models.
Take this particular gene for example:
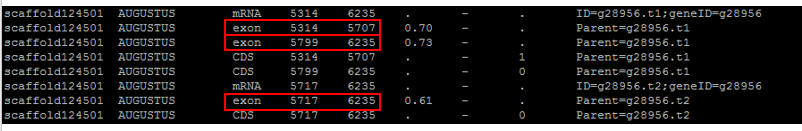
Although three exons are called, they overlap as the third is supported by a different parent transcript. The first two claim an intron while the third spans over that particular intron like this:
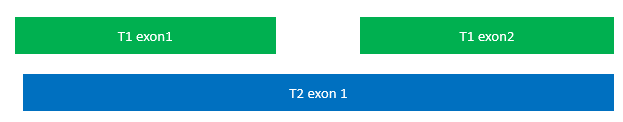
Intron prediction, when not taking into account that these are separate models of the gene space, would be wrong. To solve this, genes that show exon overlap are separated out into their own file. Then they are evaluated.
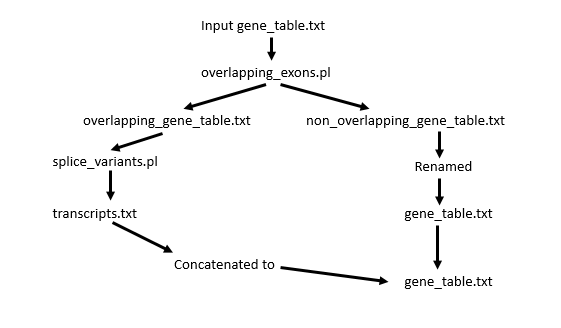
The way splice variants are confirmed is due to the labeling on the 6th column in the gene table. If an exon has a different transcript ID (often labeled t1, t2, t3…) then the exon is separated into its own “batch”. What was originally “gene22” with three separate transcripts then becomes gene22.1, gene22.2, and gene22.3.
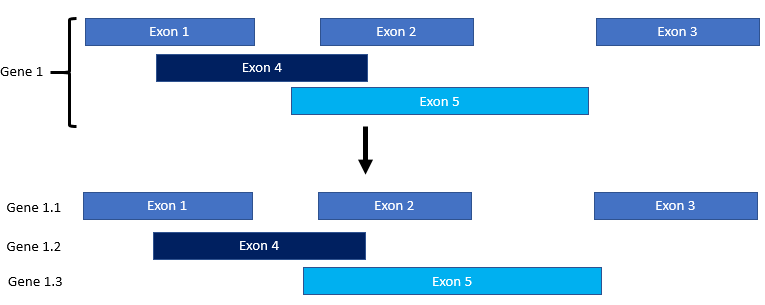
Introns are then recalculated for each of the separate isoforms. If some of the models are incomplete, they can be filtered out with other flags since they are now treated as separate “genes”. (This is because they are separated by a ### partition).
Passing genes will remain in the gene table. Results of this filter are printed in the log.
To resolve transcripts back to unique genes (selecting largest, if available, or first transcript) can be done with the --unique-genes-only flag.
Following this step, another script called task_scripts/gene_table_fixer.pl is implemented. This is done to check that exons and introns are in positive strand ascending order. Without it, printing the fastas will be problematic. It is done regardless of whether or not you use splice rescue.
--no-gene-redefining¶
The following occurs by DEFAULT. Use –no-gene-redefining to opt out. Genes that are incomplete in the annotation such that it appears they start or end with an intron are labeled as:
COMP : Complete. Note this does NOT mean there is a start and in-frame stop codon. 3*_*INC : 3’ incomplete 5*_*INC : 5’ incomplete 3*_*INC+5*_*INC : Both gene ends are incomplete
Incompleteness checks do NOT require a fasta and therefore do not judge incompleteness based on codon content. For examples of this, see the filters directly below for removing incompletes.
Genes will be trimmed to reflect available CDS in the first step following format conversion but incomplete labels will remain to identify the modification made. This is opt-out using --no-gene-redefining. All filters will be applied normally regardless of choice to trim. Note that a gene labeled as a 3’ incomplete will always fail a start codon check even if the first three nucleotides of the trimmed gene is an ATG (or alternate). Same for 5’ incompletes and stop codon checks.
--rem-3prime-incompletes¶
This option is controlled by the script task_scripts/remove_starting_introns.pl. It is designed to pull out genes that start with “intronic” space. This is almost always because of missing evidence due to missing scaffold sequence. Genes are automatically trimmed unless –no-gene-redefining is used.
Take this for example:
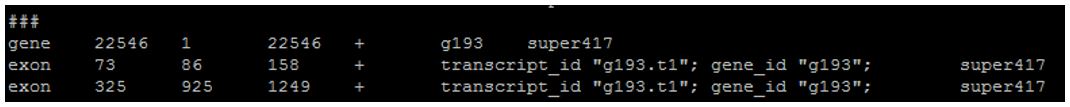
The very first exon begins 86 nucleotides into the proposed gene space. You can also see that the gene “begins” at position 1 in super417. This particular model came from BRAKER 2.0.
This script finds genes where gene start and first exon start do not agree. Passing genes will remain in the gene table. Results of this filter are printed in the log.
NOTE: This script takes into account directionality. Meaning a positive strand gene without a start intron would occur at the start of the gene (all gene coordinates are positive stranded). In a negative strand gene, missing the start intron would occur at the end of the gene.
NOTE: This filter does not take into acount sequence. To remove all incompletes with codons in mind, combine with –rem-genes-without-start-codon.
--rem-5prime-incompletes¶
This option is controlled by the script task_scripts/remove_ending_introns.pl. It is nearly identical to removing start and also takes into account strandedness and directionality. For another example in the exact same BRAKER 2.0 gene table:
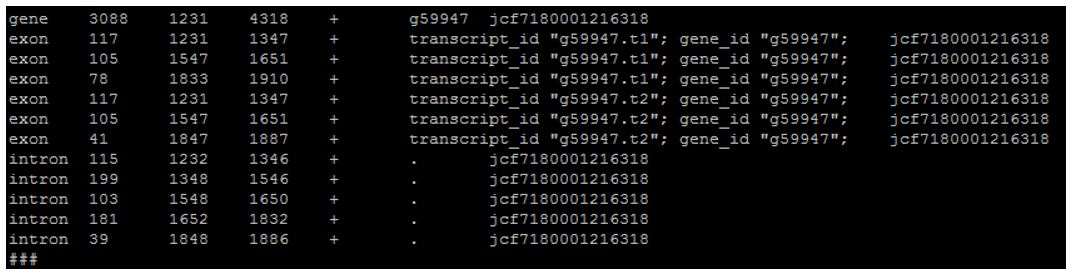
Here you can see that the last exon ends at 1887 but the gene claims to end at 4318.
Sometimes this occurs because of the scaffold ending as before, but further fasta involvement can find incomplete genes due to codon evidence.
Passing genes will remain in the gene table. Results of this filter are printed in the log.
NOTE: This filter does not take into acount sequence. To remove all incompletes with codons in mind, combine with –rem-genes-without-stop-codon.
--rem-5prime-3prime-incompletes¶
Removes genes that are BOTH 5’ and 3’ incomplete (3_INC+5_INC). Genes that are 3’ incomplete but 5’ complete and vice versa are kept in the gene table. Results of this filter are printed in the log.
--rem-all-incompletes¶
Performs the tasks of --rem-3prime-incompletes and --rem-5prime-incompletes. See above for what each task does individually. Passing genes will remain in the gene table. Results and commands of this filter are printed in the log as if each command was run separately.
--rem-monoexonics¶
Removes monoexonics based off the presence of introns. All multiexonic genes then remain in the gene table. The script that does this command is task_scripts/remove_monoexonics.pl.
--rem-multiexonics¶
Remove multiexonics based off the presence of introns. All monoexonics genes then remain in the gene table. The script that does this command is task_scripts/remove_multiexonics.pl.
--min-exon-size [number]¶
Default: 20
Creates a filter to remove genes with exons below a certain size. The script that performs this command is task_scripts/minimum_exon.pl. If you do not provide a following number, 20 is used as a benchmark for an exon that is suspiciously too small.
Passing genes will remain in the gene table. Results of this filter are printed in the log.
--min-intron-size [number]¶
Default: 20
Creates a filter to remove genes with introns below a certain size. The script that performs this command is task_scripts/minimum_intron.pl. If you do not provide a following number, 20 is used as a benchmark for an intron that is suspiciously too small. However, it might be technically possible to have introns that are less than 20 nucleotides.
Passing genes will remain in the gene table. Results of this filter are printed in the log.
--min-CDS-size [number]¶
Default: 74
Creates a filter to remove genes with a coding sequence (CDS) below a certain nucleotide length. Introns do not count, only exon sequence size. The default is based off the smallest known gene and will be used if no input is provided.
Passing genes will remain in the gene table. Results of this filter are printed in the log.
--unique-genes-only¶
This option will collapse directly overlapping genes and resolve transcripts created using -–splice-rescue.
When using --splice-rescue, multiple transcripts are created that represent the same gene. They may be isoforms of one gene or the exact same gene model repeated due to multiple pieces of evidence. Since separation treats each transcript as if it was its own gene for statistics and file-creation steps, this step will return only unique genes.
This is done by the script task_scripts/unique_genes.pl. It separates out transcripts denoted by their .1, .2, etc… modification. For those representing the same gene, the largest transcript is selected if available. Otherwise it will just take the first one.
When not dealing with transcripts, if two separate genes with different IDs share the exact same space, the first one numerically will be chosen. This only affects genes with 100% overlap where each gene is the same size and starts and ends at the same coordinates.
This step is done before any outputs are created such as statistics, fastas, output tables, or gtf files. Unique genes will remain in the gene table. Results of this filter including genes in, transcripts present, unique transcripts, non-transcript duplicates, lost, and final returned genes are printed in the log.
--sort-by-chromosome¶
This option will sort the gene table as the last step before sequence pull steps. Genes will be sorted by chromosome although genes within each chromosome will not be sorted by order. The purpose of this command is to optimize speed for sequence retrieval as switching between chromosomes can severely lag steps like fasta creation, splice site information, or codon information. This sort will be reflected in all outputs.