Fasta required flags¶
These scripts require that there is a fasta, because sequence is being evaluated. The gFACs.pl script will index your fasta, and then task scripts that require sequence will find and use that index. If there is already an index, the indexing step will be skipped.
To specify a fasta:
--fasta /path/to/your/nucleotide/fasta.fasta¶
Bioperl will create an index with the ending “.fasta.idx”. It is a fairly fast process. The file may end with .fa or .fasta, but no other naming formats can be recognized. gFACs will also check to ensure the scaffold names in the genome match the input annotation.
NOTE: This fasta MUST MUST MUST be the same fasta used when making your particular gff3/gtf/gff. Bioperl needs to recognize the name on the fasta info line to the sixth column in the gene table.
--canonical-only¶
Analyzes introns for a canonical splice sites (GT-AG on the positive strand). The script that performs this task is task_scripts/canonical_only.pl.
To pass, all introns in a gene must have canonical splice sites. Monoexonics will also pass this filter because they do not have the evidence to be pulled out. Of course, monoexonic genes can be removed by –rem-monoexonics filter.
Genes that pass this filter are kept in the gene table and results are printed in the log.
NOTE: Splice sites take into account directionality and reverse compliment.
--rem-genes-without-start-codon¶
The first three nucleotides of the sequence are analyzed to match ATG. With this flag alone, no alternate start codons are taken into account (use --allow-alternate-starts to include alternate starts). This task is performed by task_scripts/rem_genes_without_start.pl. Again, gene directionality is considered.
NOTE: Genes marked 5’ incomplete are assumed NOT to have a start codon and are removed regardless if the gene starts with a Met.
Genes that pass this filter are kept in the gene table and results are printed in the log.
--allow-alternate-starts¶
If --rem-genes-without-start-codon is used, the start codons of GTG and TTG will also be included alongside ATG. This may be useful in Prokaryotic annotations. This task is performed by task_scripts/rem_genes_without_start_alternate.pl.
Outputs are identical to --rem-genes-without-start-codon.
--rem-genes-without-stop-codon¶
The last three nucleotides of the sequence are analyzed to match TAA, TAG, and TGA. Currently, all end codons are assumed to be within the reported gene. This task is performed by task_scripts/rem_genes_without_stop.pl. Again, gene directionality is considered.
Following this step, a script called task_scripts/frame_detection.pl is run. It is designed to pick out any genes that technically have a stop codon as the last three nucleotides, but it is not real because the codon is actually out of frame. These are rare occurrences, often happening on negative strand genes that run into the beginning of a scaffold where the first three nucleotides of the scaffold are a reverse complement stop codon. To solve this, any gene whose CDS is not divisible by 3, is removed.
NOTE: Genes marked 3’ incomplete are assumed NOT to have a stop codon and are removed regardless if the gene has a terminating in-frame stop.
Genes that pass this filter are kept in the gene table and results are printed in the log.
--rem-genes-without-start-and-stop-codon¶
Removes genes that lack BOTH a start and stop codon. Genes that have a start codon but no stop and vice versa are kept in the gene table. Results of this filter are printed in the log.
--allowed-inframe-stop-codons [number]¶
Default: 0
Creates a filter that removes genes based on the presence of a stop codon that is not the last codon in the gene. For example, setting this parameter as 1 will allow one other stop codon between the methionine and the terminating stop codon.
If you are not filtering for start and stop codons, this will still work so long as there are stop codons within the amino acid sequence but not necessarily at the end.
Genes that pass this filter are kept in the gene table and results are printed in the log.
--splice-table¶
To understand splice usage, a splice-site table is printed to the log that tells the frequency of every type of splice site used. This command is performed by task_scripts/splice_table.pl.
The splice table will look something like this:
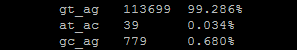
This splice table will show you everything present in the file adjusted to lower case letters including N-bases. If you specify canonical genes only, the table will only show you gt_ag counts.
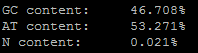
--nt-content¶
The CDS (all exon sequences) is analyzes for GC, AT, and N content by percent composition. This information is printed to the log. Here is an example of the output: